Welcome to the User Manual for REDCRAFT!
1. Introduction
REDCRAFT is an open-source software tool for determining a protein's structure using residual dipolar couplings (RDCs). It allows simultaneous determination of a protein's structure and dynamics. Its effectiveness has been demonstrated on both synthetic and experimental data. REDCRAFT contains stages that allow the incorporation of user-specified dihedral angle constraints, such as those produced by TALOS or a restriction to specific regions of Ramachandran space. It is robust with respect to noise and missing data. The program is highly efficient and can produce a structure for an 80-residue protein within two hours.
Originally started by Dr. Homayoun Valafar the REDCRAFT project has had a number of contributors including
- Mike Bryson
- Paul Shealy
- Zach Swearingen
- Mikhail Simin
- Casey Cole
- Earron Twitty
For questions and comments please feel free to contact Casey Cole.
2. Installation
Packages are available for Windows, MAC and Linux.
Download current package at --> http://ifestos.cse.sc.edu/software.php#redcraft
- Navigate to where the redcraft package was downloaded and saved (probably your Downloads folder)
- cd /Downloads
- Become sudo user :
- su or sudo su
- Move package to location of installation :
- mv redcraft.tar.gz /opt
- cd /opt
- Untar/unzip the file. This step will create a subdirectory named redcraft containing all of the files.
- tar -zxf redcraft.tar.gz
- Although this package comes with the executable files, you may wish to compile the program specifically for your environment. You can compile and install REDCRAFT from the redcraft directory using the make functionality:
- make
- make install
3. Data Preparation
The current version of REDCRAFT accommodates the analysis of six types of RDC data (illustrated in Table 1.) from an arbitrary number of alignment media. In addition to the RDC data, REDCRAFT takes advantage of the residue type and scalar coupling data as shown in Table 1.
| Data type | Atoms involved |
|---|---|
| Residue type | Residue corresponding to Ciα of residue i |
| Three bond J-scalar coupling | Hia - HiN |
| RDC | C(i-1) - Ni |
| RDC | Ni - HiN |
| RDC | C(i-1) - HiN |
| RDC | Ciα - Hiα |
| RDC | Hiα - HiN |
| RDC | H(i-1)α - HiN |
Table 1: A list of all the data that comprise the input to REDCRAFT.
Note that successful analysis of structure and motion does not necessarily require all of the above data for all of the peptide planes. In fact, there is no concrete study alluding the minimum amount of data requirement. The above data for each peptide plane from each alignment medium can be collected into a single file in blocks as described below:
General Format for REDCRAFT fileAA J-Coupling Comment
N-C error
N-H error
H-C error
CA-HA error
HA-H error
H-HA error
A value of 999 for any entry (other than the confidence factor and the amino acid type) is considered to be a missing value. The following example illustrates the content of an RDC file for the first three residues of a protein. Note that values of 999 indicating missing RDC data. Often time, it is useful to provide a meaningful comment.
ExampleRDC.1
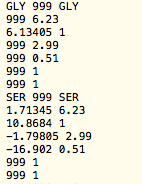
The RDC data files need to be specifically named RDC.? where ? denotes an integer number corresponding to different alignment media. For example, when RDC data are available from two alignment media, the program expects to find RDC.1 and RDC.2 files that contain the respective RDC data.
How can I get RDC data?- Experimental Data
- Calculated Data
- REDCAT : open source engine to back calculate RDCs from a PDB file. Complete with an easy-to-use GUI and robust features, providing an easy way get RDCs from nearly any protein structure.
- REDCAT2REDCRAFT.prl : converts a REDCAT file to REDCRAFT file
- excel-REDCRAFT.prl : converts a csv file generated from a spreadsheet to REDCRAFT file
- XplorTorsion2REDCRAFT.prl : converts an Xplor torsion angle file to REDCRAFT file
The first stage of structure determination is based on eliminating torsion angles incompatible with Ramachandran space and J-coupling. Finally, the surviving torsion angles are ranked on the basis of fitness to the RDC data available from the juxtapositional peptide planes. For more detail please refer to the following articles [1-3]. The script stage1 is located in the scripts directory and can be used to perform the Stage-I analysis as follows:
stage1 <RDC prefix> [Ramachandran space [GLY-ramachandran space [RDC RMSD cutoff]]]- Prefix of your data files: Required
- Each alignment medium data should be in its own file with the same file prefix ending with .1, .2 etc... stage1.prl needs to know the prefix (such as RDC if your file names are RDC.1 and RDC.2) It will find the count of RDC files automatically.
- Ramachandron filter for non-GLY residues : optional
- 1 (default, more strict)
- 2 (less strict)
- all (entire space)
- Value of 1 is the most restrictive Ramachandran space. The torsion angle clusters for beta-sheet and alpha-helicies do not touch.
- Ramachandron filter for GLY : optional
- 0 (default, GLY Ram. space)
- 1 (less strict)
- all (entire space)
- Glycines are known to have a greater freedom of variation due to their lack of side chain. This is important to consider these extra possible torsion angles. Value of 0 utilizes a lesser restrictive space common for GLY residues. Value of 1 is the same restriction as non-glycine space.
- Cutoff (in Hz) : optional
- any decimal value
- skip
- stage1 ranks and sorts each torsion angle combination based on its local fitness to available RDC data. The user can specify a cutoff fitness after which torsion angles are discarded. By default no values are discarded. A keyword skip will tell stage1 not to compute the fitness at all. This does not hinder the quality of stage2, but may inconvenience further analysis. Skipping the fitness greatly speeds up stage1.
Stage-II
Stage-II uses the ranked lists of the surviving local geometries (from Stage-I) to create and extend the fragment that most optimally fits the experimental data. Stage-II begins with the starting peptide plane and continually adds on peptide planes until the ending residue has been reached. Each new list of predicted structures is ranked after each new peptide plane is added.
Stage-II looks for a configuration file redcraft.conf before proceeding with protein folding. To create a new generic configuation file run the command stage2 --create-new. It will create a file named redcraft.conf.
Example redcraft.conf file -->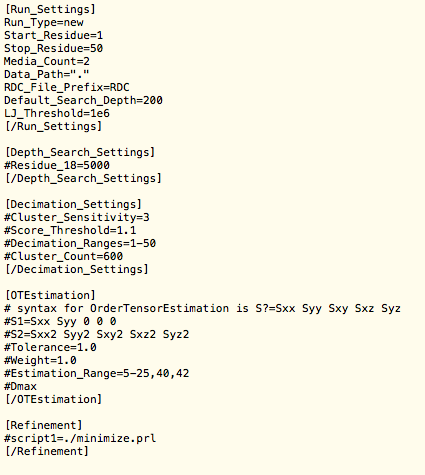
* Notice that a "#" at the beginning of a line indicates a comment. This means that this line of the file and it's corresponding feature is disabled. To enable the line, simply remove the "#".
The first block [Run_Settings] will be the core for your REDCRAFT run. It includes the following parameters :
- Run_Type {new | continue} :
- new will start a run with 1 residue from residue number Start_Residue until Stop_Residue
- continue allows you stop a run and pick up where you left off at a later time
- Start_Residue {numerical, 1 or greater} :
- This value specifies a starting point for angle files from Stage-I and RDC data. (Useful for fragmented folding)
- Stop_Residue {numerical} :
- This parameter is the last residue number that will have predicted torsion angles. It can be any residue number that is less than or equal to the last residue in the RDC data files.
- Redcraft also enables reverse folding, which allows you to swap the start and stop residue. A tutorial demonstrating this feature is available in the tutoral section.
- Media_Count :
- This is the number of alignment media created for the protein; we shall call this parameter m. The alignment media must have the names Prefix.1, Prefix.2, etc. They must always be numbered in order from 1 to m. See the section on Data Generation to see how to create these files.
- Data_Path :
- Path to the data sets you are using
- Default is "." indicating that your data is in the same directory that you plan to run the program from
- RDC_File_Prefix {string data file prefix} :
- The datafiles containing the RDC information do not necessarily have to be named RDC.1, RDC.2 etc. This field allows a custom prefix. Stage-II will expect the data files to be in format of Prefix.1, Prefix.2 etc.
- Default is RDC (so if you have 3 data sets your file names would be RDC.1, RDC.2 and RDC.3)
- Default_Search_Depth {numerical, greater than 0} :
- This parameter refers to the depth of search and it is denoted as d. When extending a fragment by adding a peptide plane, the lists of angles are combined to create a large list (usually at least 10,000 angle combinations). REDCRAFT will take the top d angle combinations and eliminate the rest. The larger the depth of search, the better the results. However, a greater depth of search will increase computation time. We recommend a typical search depth of 2000. Deeper search depths may be required for more corrupt data, or lack of data.
- Default is 200
- LJ_Threshold
- Lennard-Jones Threshold is discussed in depth in section 5
- Default is 50
Features :
- Search Depth
- Decimation
- L-Minimization
- OTEstimation
- Collison Detection
6. User Interface
Coming soon...
7. Useful Scripts included with REDCRAFT
Redcraft has a ton of PERL scripts that aim to aid this software's analysis, and interpretation of its results.
Some of these scripts are described below. If you have any questions concerning these or other supplied scripts please contact the current lead developer. All scripts are written in Perl and can be found in the /scripts subdirectory distributed with the newest version of the REDCRAFT software package.
Data Conversion
- REDCAT2REDCRAFT.prl
- Converts a REDCAT file to REDCRAFT file
- Usage: perl REDCAT2REDCRAFT.prl <list of PHI angles> <REDCAT file>
- The list of PHI angles is needed to calculate the J-coupling for each residue and should be formated as such -->

[AA PHIangle]
- excel-REDCRAFT.prl
- Converts a csv/txt file generated from a spreadsheet to REDCRAFT file
- Usage: excel-REDCRAFT.prl <input file> <output file>
- The script expects a space delimited file in the general form where each line represents RDC data from each residue -->
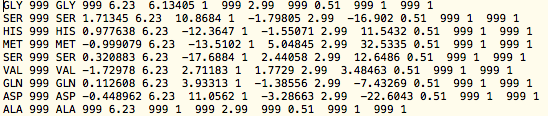
[AA J-coupling comment CN error NH error CH error CaHa error HHa error HaH error]
- CreateAngles.prl
- Creates ".angles" files for each specified angle pair in the argument
- Usage: CreateAngles.prl <phi1> <psi1> [<phi2> <psi2>[<phi3> <psi3>...
This script needs to be configured by editing the actual file, since its arguments have an unconditional use. The first 4 variables mentioned in the script are its only configuration.
$SKIPFIRST -- 1 or 0. Should the first PHI angle be skipped in the permutation? (in case no data affects the first PHI) $SKIPLAST -- 1 or 0. Should the last PSI be skipped? $grid -- What should the step size be between various values? REDCRAFT's default files use 10 degree grid. $padding -- How far from the specified angles should the divergence be?
- Filters out angles from a .angles file that are not within a radius of a phi-psi pair
- Usage: FilterPhiPsi.prl <.angles file> <phi> <psi> [radius]
- Adjusts the RDC vector weight (error contribution) based on contribution of other vectors (even in multiple alignments)
- Usage: multiweight.prl <RDC input prefix> <RDC output prefix> <media count>
- Prunes a given RDC file of specified RDC vector type with an optional residue name restriction
- Usage: RemoveRDC.prl <input RDC file> <output RDC file> J 1 2 3 4 5 6 [Residue name]
- The J 1 2 3 4 5 6 is a boolean mask of which vector types should be deleted.
- Ex. To remove just the J-coupling use the mask : 1 0 0 0 0 0 0
- Ex. To remove everything except NH use : 1 1 0 1 1 1 1
- Fragments.prl
- Displays the RDC data density for a given RDC prefix
- Usage: Fragments.prl <RDC prefix> [Media Count]
Example output:
Res Phi Psi 1 3 2 2 3 2 3 3 2 4 3 2 5 3 2 6 3 2 7 1 1 8 0 0 9 1 1 10 3 2 11 3 2 12 2 2 13 3 2 14 2 1 15 3 2
- Prints a correspondence between the residue number, RDC fitness, and BB RMSD of the best structure in the fragment
- Usage: CalcBBRMSD.prl <start residue> <stop residue> <reference pdb file>
When a reference PDB structure is available this tool allows you to create a 3 column list of relationship data:
ResidueID RDC-RMSD BB-RMSD
The RDC-RMSD and BB-RMSD are of the structure with best-fit RDC RMSD for this iteration. REDCRAFT does not produce a result for the first iteration, so for a segment from 1 to 10, the first result would be 2.pdb. An example of observed CalcBBRMSD.prl run would be:
$ CalcBBRMSD.prl 2 15 reference.pdb 2 1.01754e-15 1.409 3 7.48886e-16 1.313 4 3.53271e-16 2.569 5 8.66401e-16 2.877 6 0.0109947 2.321 7 0.0423316 2.135 8 0.0516006 2.699 9 0.071958 3.954 10 0.640688 0.247 11 0.680786 0.235 12 0.682075 0.245 13 0.678333 0.287 14 0.679285 0.315 15 0.682678 0.316
9. References
[1] Bryson M, Tian F, Prestegard JH & Valafar H. Redcraft: a tool
for simultaneous characterization of protein backbone structure and
motion from rdc data. J. Magn. Reson. (2008) 191: pp. 322-334.
[2] Valafar, H., Simin, M., & Irausquin, S. (2012). A Review of
REDCRAFT: Simultaneous Investigation of Structure and Dynamics of
Proteins from RDC Restraints. Annual Reports on NMR Spectroscopy, 76,
23–66. doi:10.1016/B978-0-12-397019-0.00002-9
[3] Shealy P, Simin M, Park SH, Opella SJ, Valafar H, Simultaneous
Structure and Dynamics of a Membrane Protein using REDCRAFT:
Membrane-bound form of Pf1 Coat Protein, Journal of Magnetic Resonance,
2010 Nov;207(1):8-16. Epub 2010 Jul 30, PMID:20829084
Going on in our lab :
- REDCAT
- msTali
- 2DPDPA